Single-cell RNA sequencing (scRNA-seq) enables the analysis of gene expression in individual cells, unveiling new subsets of cells in different states or diseases through statistical analysis. Despite the capability of current scRNA-seq techniques to characterize thousands of cells in a single experiment, they only capture a small fraction of the transcriptome (10-15%), rendering rare or low-abundance genes undetectable. To address this issue, Dr. Biton and his team have developed a novel scRNA-seq approach that enables the detection of a targeted set of genes of interest that are typically overlooked, in addition to the 10-15% of the transcriptome captured by conventional methods. This gene set can be tailored to cover specific pathways like inflammation, providing a more detailed view of the transcriptional profile of a single cell. The technique is highly sensitive, accurate, efficient, high-throughput, and cost-effective.
scRNA-seq enables the detection of gene expression in individual cells, uncovering distinct cell subpopulations across various states or pathologies, providing valuable insights into disease diagnostics, progression and response and allowing the identification of potential targets for therapy. Modern scRNA-seq can analyze numerous transcripts from thousands of cells simultaneously, clustering cells by shared gene expression patterns to profile cell populations and states, revealing the full cellular diversity in a sample.
The most commonly used scRNA-seq methods include 10x Genomics 25 Chromium, Smart-seq2 (SS2), Mars-seq, and CEL-seq2, each designed to address specific biological questions and utilizing either microfluidics or well-based platforms. Microfluidics-based methods, such as 10x Chromium, enable the simultaneous sequencing of thousands of cells, but require freshly isolated tissue samples, making long-term experiments difficult. In contrast, well-based methods, including Mars-seq, SS2, and CEL-seq2, allow long-term storage of single cells in well plates until samples are collected and processed together to reduce batch effects, but have limited throughput capacity, making them costly and time-consuming. Multiplexing, through cell-specific barcoding, can enhance the throughput ability of well-based methods to match that of microfluidics-based methods.
Despite recent advancements, scRNA-seq only captures approximately 10-15% of a cell's transcriptome, indicating a need for improved and cost-effective single-cell RNA sequencing and analysis technologies.
Dr. Biton and his team have created a distinctive scRNA-seq technique that allows for the identification of a specific group of genes that are typically overlooked in standard methods, in addition to the 10-15% of the entire transcriptome. These genes can be selected to encompass particular pathways, such as inflammation
A new method called Well-based RNA Amplification and Pooling (WRAP-seq) was developed by the team. The WRAP-seq library preparation process involves four simple reactions: reverse transcription, amplification, tagmentation, and 3' product selection. One significant advantage of WRAP-seq is that the entire library preparation can be completed in a single day, which saves time and reduces the amount of labor required. WRAP-seq incorporates cell barcodes for pooling libraries, allowing for the processing of multiple libraries simultaneously, reducing batch effects and increasing throughput. Furthermore, WRAP-seq is relatively inexpensive due to its multiplexing capability. To ensure accurate transcript quantification, WRAP-seq libraries have unique molecular identifiers (UMIs).
In conjunction with whole transcriptomic analysis, WRAP-seq is combined with targeted sequencing (TRAP-seq) to detect low-abundance genes of interest, such as transcription factors and cytokines. This tool enhances sequencing resolution and provides a comprehensive analysis of the immune compartment that cannot be achieved with current methods.
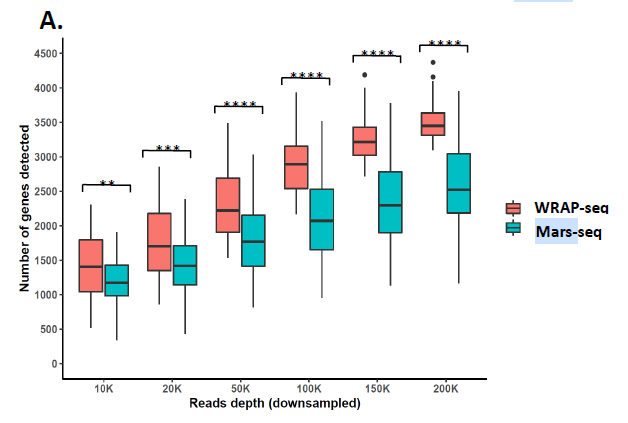
Figure 1 – the sensitivity of WRAP-seq is superior to the MARS-seq.
- Single-cell sequencing of targeted genes of interest in parallel to whole transcriptomics analysis
- Superior sensitivity compared to current solutions (Mars-seq)
- Efficient High-throughput
- Cost-effective
- Accurate quantification
The method is fully developed. As proof of concept, the team sequenced WRAP-seq libraries prepared from HEK293T cells and compared WRAP-seq’s sensitivity with Mars-seq, using a previously published Mars-seq dataset of HEK293T cells, demonstrating the superiority of WRAP-seq (Figure 1).

