Trinucleotide repeats expansion disorders (TREDs) are a set of diseases in which repetitions of short DNA segments (three nucleotides) occur and accumulate in the genome. This is the cause of several neurological and neuromuscular disorders such as Huntington's disease (HD) and amyptrophic lateral sclerosis (ALS), which are lethal and currently incurable. The current technology offers a set of pharmacological compounds that can inhibit the transcription of only the mutated expanded allele, without affecting the wt allele,thus potentially treat neurological pathologies caused by TREDs such as HD.
TREDs are a group of human diseases in which repeats of three nucleotides accumulate in the genome. Once the number of repetitions crosses a threshold, they become unstable, which can result in defects in the protein encoded by the gene, changes in the regulation of gene expression, toxic RNA products, or chromosome instability. In general, the larger the expansion the faster the onset of disease and its severity.. These repeats are hallmarks of several inherited degenerative neurological disorders including HD, ALS, and Frontotemporal Degeneration (FTD), which are all lethal and currently incurable. A key protein involved in the transcription of the mutant trinucleotide repeats genes, but not the normal gene, is the transcription factor Spt5 along with its partner Spt4. Spt4/Spt5 play an important role in expression of the mutant gene by interacting with RNA polymerase II (the molecular machinery that transcribe genes) and enabling transcription of long trinucleotide repeats. Therefore, methods of specifically modulating such a target would have significant applications.
The group of Prof. Rivka Dikstein identified small molecules that specifically inhibit the association between RNA polymerase II and Spt5, resulting in inhibition of the expression of the mutated Huntington gene (Htt), without affecting the normal allele.
The Dikstein group has developed a high throughput assay to screen small molecules that inhibit the Spt5 – RNA polymerase interaction. The group screened 100,000 small molecules and narrowed them down to 18 biologically active compounds named Spt5-Polymerase Inhibitors (SPIs). These SPIs were then tested in striatal (neural) cell lines derived from knock-in mice in which the first exon of the mouse Htt gene was replaced with either a wild type human 7xCAG trinucleotide repeat (Q7), or a mutant 111xCAG trinucleotide repeat (Q111). Upon in vitro exposure to SPIs, striatal cell lines exhibited a reduction of Q111 Htt expression relative to Q7 Htt (Fig 1), demonstrating that the inhibition caused by the SPIs could differentiate between normal and aberrant genes.
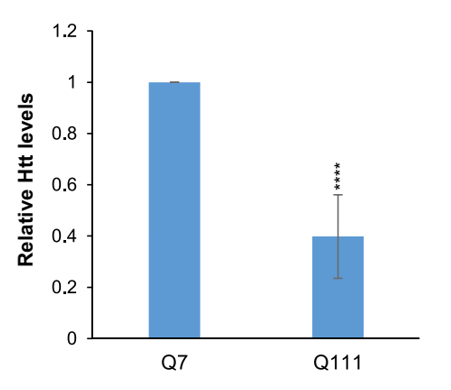
Figure 1: WT Q7 and Mutant Q111 Htt protein levels in cells normalized to β-actin, with Q7 Htt levels set to 1. Error bars represent the means ± SEMs of at least 3 independent experiments, with the asterisks denoting statistically significant differences relative to Q7. (Bahat et al., 2019, Molecular Cell)
- Potential small molecule therapeutics for the treatment of HD
- SPIs could also potentially be used for the treatment of other TREDs, such as ALS, FTD, myotonic dystrophy type 1, and others
The group of Prof. Dikstein has performed rigorous work to screen for small molecules discovering an initial set of biologically active SPIs. Activity was confirmed in a number of in vitro assays. Currently, in vivo experiments are conducted in HD mouse models to assess the efficacy and toxicity of the compounds. Preliminary In vivo results revealed no toxic effect of the drugs in the highest concentration tested, and suggest improvement in motor activity and anxiety. Future plans include further medicinal chemistry to improve the most promising SPIs, and testing these SPIs in an ALS mouse model.
Bahat, Anat, Or Lahav, Alexander Plotnikov, Dena Leshkowitz, and Rivka Dikstein. 2019. "Targeting Spt5-Pol II by Small-Molecule Inhibitors Uncouples Distinct Activities and Reveals Additional Regulatory Roles." Molecular Cell 76 (4): 617–31.e4.

